Examples
Video clips
These short clips should give just a feel for how ASCIIGenome works in interactive mode.
This example loads a bam file and shows the read track at the bottom and the coverage track on top. After loading the bam file, the follow operations are executed at the command prompt:
Go to specified region.
Zoom in.
Repeatedly move forward by half a window size.
Zoom out.
Filter reads using samtools-like syntax.
Here there are two bigWig files of ChIP-Seq profiles. The corresponding peak regions are separately loaded. The following operations are executed at the command prompt:
Change track height to 12 lines each.
Add two narrowPeak files, captured with the glob
*.narrowPeak.gz.Re-order tracks.
Zoom out.
Hide title lines.
Change limits of the Y-axes to be from 0 to maximum of all tracks.
Move to the next bed feature.
Show help for the
ylimcommand.
Why the command line
At first it may be nonsensical to use a viewer without graphical interface. In addition, typed commands involve a steeper learning curve and more frustration. However, the command line interface has some great benefits which, at least in part, explain why most bioinformaticians prefer R, python and Unix tools over Excel and Galaxy (which are great, by the way, just like IGV is great). Some advantages of the command line interface over the GUI:
Streamline repetitive tasks.
Finer control of the commands.
Self-documented and therefore reproducible.
This example should illustrate these points. For a start, most of my data files, especially the big ones, live on the institutes’s computer cluster or on our group server. Almost nothing is stored on my workstation. Consequently pretty much all the work I do is via Unix commands, bash and the familiar samtools, bedtools, etc. Popping up a GUI is often a disturbance of the workflow so having a visualisation tool with the same interface (i.e. command line) as these tools is more natural.
Now, say we want to visualise the following files:
ts058_TS10-PEO1-Pt-A2.tdf
ts059_TS10-PEO1-Pt-A4.tdf
ts060_TS11-PEO1-DMF-A13.tdf
ts061_TS11-PEO1-Pt-A12.tdf
ts062_TS11-PEO1-Pt-A7.tdf
ts063_TS11-PEO4-DMF-A5.tdf
ts064_TS11-PEO4-Pt-A2.tdf
ts065_TS11-PEO4-Pt-A4.tdf
ts069_PEO1-DMF-A5.tdf
ts070_PEO1-Pt-A18.tdf
ts071_PEO1-Pt-A2.tdf
Loading all these files by clinking one by one through a GUI can be annoying especially if they are in different directories, not counting the time spent to pop up the GUI and scroll through the relevant menus. With ASCIIGenome you can probably just do one of the following:
ASCIIGenome ts0{58..71}*.tdf
ASCIIGenome *PEO1*.tdf *PEO4*.tdf
ASCIIGenome `find . -name '*PEO*.tdf'` ## If files are in different subdirs
ASCIIGenome ts058_TS10-PEO1-Pt-A2.tdf ts059_TS10-PEO1-Pt-A4.tdf <etc>
The command line you have used can be copied in your documentation for reference and it can be used again by copying and pasting it to the terminal.
Once these files have been loaded you may want to order them to have the PEO1 tracks before the PEO4s. This is just:
orderTracks PEO1 PEO4
Similarly, settings can be changed without the need of scrolling through menu options, for example:
colorTrack blue PEO1 <- Turn blue the PEO1 tracks
trackHeight 10 DMF <- Make 10 lines high the tracks matching DMF
Furthermore, the commands issued at the prompt can be scrolled with the UP and DOWN arrow
keys. So if we want to change the colour of the PEO1 tracks again we just need to press UP two times,
bring back the colorTrack blue PEO1 command, and edit it as required.
We can put this together in a single command which, again, can go to the documentation:
ASCIIGenome -x 'orderTracks PEO1 PEO4 && colorTrack blue PEO1 && trackHeight 10 DMF' ts0{58..71}*.tdf
Open and browse
Open some peak and bigWig files from ENCODE.
Note
Opening remote files is a little slow (IGV seems equally slow). You might also need to start Java with the option -Djava.net.useSystemProxies=true (see also issue#6)
encode=http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhTfbs
ASCIIGenome \
$encode/wgEncodeSydhTfbsGm10847NfkbTnfaIggrabPk.narrowPeak.gz \
$encode/wgEncodeSydhTfbsGm10847NfkbTnfaIggrabSig.bigWig \
$encode/wgEncodeSydhTfbsGm12892Pol2IggmusPk.narrowPeak.gz \
$encode/wgEncodeSydhTfbsGm12892Pol2IggmusSig.bigWig
Find the first feature on the first file, then change colour of one of the tracks. Reset y axes to span 0 to 50, finally save as pdf to default file name:
[h] for help: setGenome hg19
[h] for help: next #1
[h] for help: colorTrack pink wgEncodeSydhTfbsGm12892Pol2IggmusSig
[h] for help: ylim 0 50
[h] for help: save %r.pdf
Result on terminal screen should look like this:

The screenshot is saved to chr1_996137-1003137.pdf, note that the variable %r is expanded to the genomic coordinates.
Finding & filtering stuff
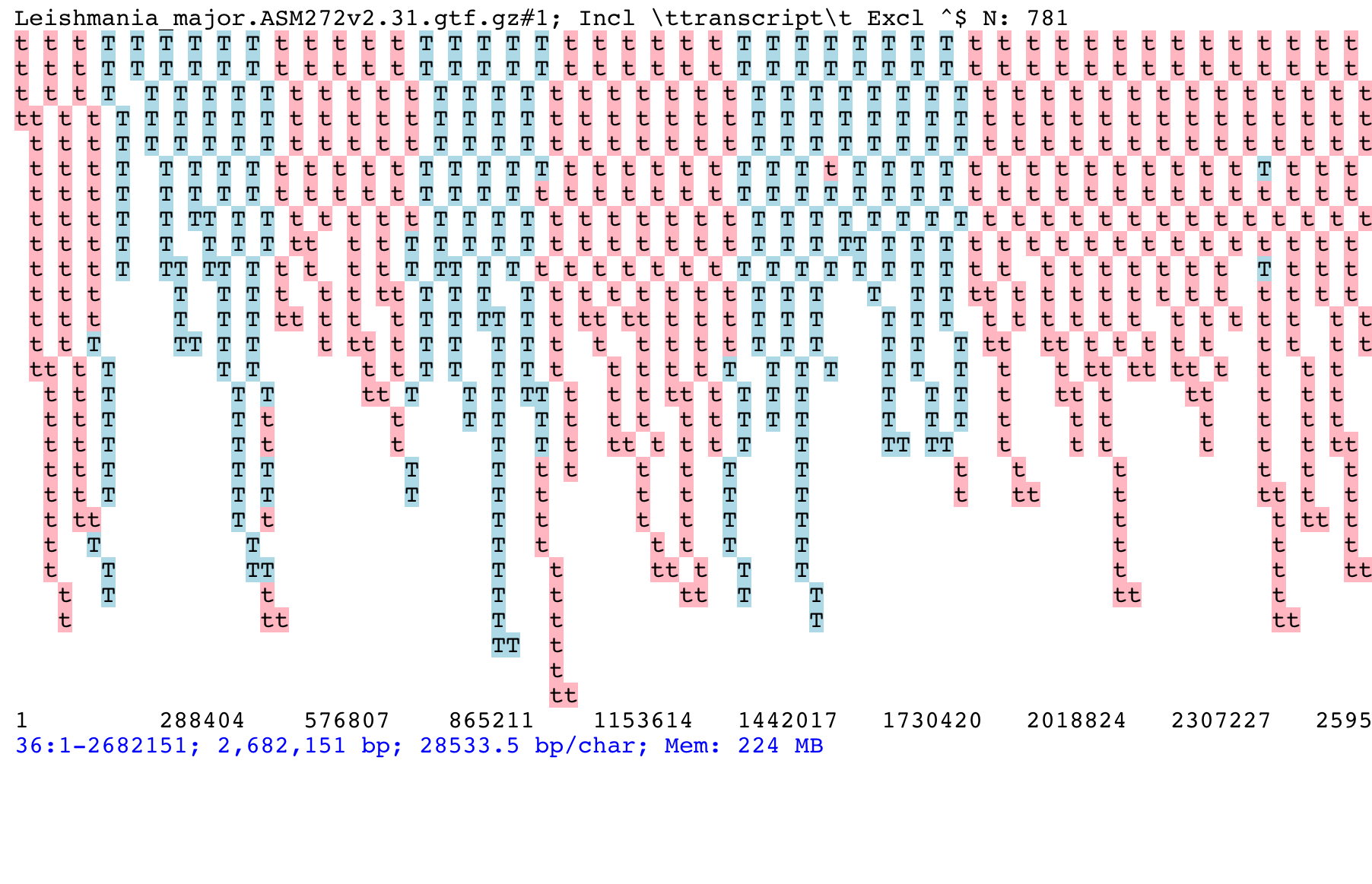
Once started, ASCIIGenome makes it easy to browse the genome. The picture below shows the distribution of transcripts on chromosome 36 of Leishmania major. It is clearly visible how transcripts in Leishmania tend to be grouped in blocks transcribed from the same direction (blue: forward strand, pink: reverse strand). Note how overlapping features are stacked on top of each other.
This screenshot has been produced by first loading the L. major GTF file:
ASCIIGenome ftp://ftp.ensemblgenomes.org/pub/release-31/protists/gtf/leishmania_major/Leishmania_major.ASM272v2.84.gtf.gz
At the command prompt issue the following commands:
[h] for help: goto 36:1-2682151
[h] for help: grep -i transcript
[h] for help: trackHeight 100
Now return to the start of the chromosome and find the first feature containing LmjF.36.TRNAGLN.01, print it to screen:
[h] for help: 1
[h] for help: find LmjF.36.TRNAGLN.01
[h] for help: print
Now showing:

Advanced filtering with awk
From version 1.5.0, the awk program is available within ASCIIGenome to filter interval features and SAM records. See the command reference for awk for more detials. This an example of using awk to filter reads containing deletions, i.e. where the cigar string contains the character D:
awk '$6 ~ "D"' *.bam
The syntax $6 selects the 6th column, the one containing the cigar string, the operator ~
returns true if the cigar string contains the string D. As usual, the last positional argument
applies the command to the tracks matching the given regex.
The internal function get() returns the value of the additional tags in SAM, GFF/GTF, or VCF files.
For example, to select only alignment containing mismatches, i.e. where the NM tag is greater than zero, use:
awk 'get("NM") > 0'
Filter VCF records where tag CalledBy contains “WCMC”. Note the ~
operator to perform refular expression matching:
awk 'get("CalledBy") ~ "WCMC"'
or where CalledBy does not contain WC:
awk 'get("CalledBy") !~ "WCMC"'
If CalledBy contains a list of values you can access them with the second
parament of get(tag, index) indicating the index of the value to be returned, 1-based.
E.g., return records where the second element of CalledBy equals
BCM:
awk 'get("CalledBy", 2) == "BCM"'
When working with long reads, it is often useful to filter for aligmments longer than a cutoff:
awk 'getAlnLen() > 2000'
Batch and non-interactive mode
ASCIIGenome can be integrated in a script to be executed without direct human intervention. For example, a simple bash script may contain the following commands:
#!/bin/bash
## Find ChIP-Seq peaks
CHIP=ChIP.bam
macs2 callpeak -t $CHIP -c input.bam -n out
## Output pdf in a "control" region for later visual inspection
ASCIIGenome -ni -r chr1:1000000-1020000 -x "save ${CHIP}.ctrl.pdf" \
$CHIP input.bam out_peaks.narrowPeak > /dev/null
In this script, a ChIP-Seq sample is first analysed to find peaks against an input control. ChIP, input and output from the peak caller are then loaded in ASCIIGenome to visualize a region of interest. ASCIIGenome will save the image in pdf file named after the ChIP sample and exit. An investigator can later inspect the pdf figure to assess the quality of the ChIP or to check whether a peak has been detected.
The example above can easily be extended to several regions to be visualised in
batch for one or more tracks. For example, you have a list of ChIP-Seq peaks or
RNA-Seq genes and you want to see the coverage profiles together with an
annotation file. ASCIIGenome allows batch processing via the
--batchFile/-b option.
This script iterates through the intervals in peaks.bed. For each interval, it displays two
bigWig, a gtf file and the peak file itself. Each interval is zoomed out 3 times and the screenshot
saved as pdf to /tmp/peak.%r.pdf, where %r is a special variable expanded to the current
coordinates as chrom_start-end.:
ASCIIGenome -b peaks.bed \
-x 'zo 3 && save /tmp/peak.%r.pdf' \
chipseq.bigwig \
input.bigwig \
gencode_genes.gtf \
peaks.bed > /dev/null
To save all the screenshots in a single pdf use the >> operator in the save command, e.g. save >> myScreenshots.pdf.
Finding sequence motifs
The reference fasta sequence can be searched for sequence motifs specified via regular expressions or via IUPAC notation.
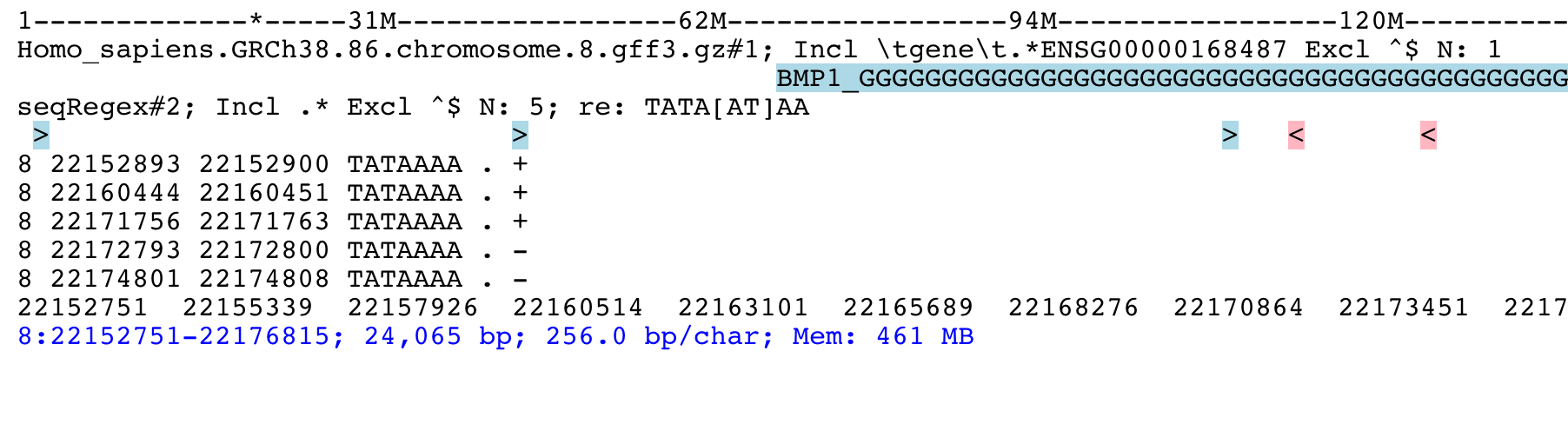
This example is from Biostars. We want to find matches of the motif TATAWAA near gene ENSG00000168487.
First load the reference sequence and a (remote) annotation file:
ASCIIGenome -fa Homo_sapiens.GRCh38.dna.chromosome.8.fa \
ftp://ftp.ensembl.org/pub/release-86/gff3/homo_sapiens/Homo_sapiens.GRCh38.86.chromosome.8.gff3.gz
Then at the command prompt issue these commands:
find ENSG00000168487
grep -i \tgene\t.*ENSG00000168487 gff3
seqRegex -iupac TATAWAA
zo 8
print seqRegex
print seqRegex > matches.bed
save matches.png
Explained: Find the gene ENSG00000168487, for clarity only show the “gene” feature (grep...).
Then search the motif TATAWAA interpreted as iupac notation; zoom out x times (e.g. 8 times) to see some
matches in the sequence.
The matches here are shown on screen with print seqRegex and then saved to file with print seqRegex > matches.bed. Finally save a picture as png, shown here: